今天给大家介绍一篇由Anukrati Goel等人合作的,于前段时间发表在Food Chemistry的一篇文章,文章中作者介绍了一种基于计算机驱动的方法,可以从大型天然化合物数据库中筛选出潜在的新型甜味剂。
今天给大家介绍一篇由Anukrati Goel等人合作的,于前段时间发表在Food Chemistry的一篇文章,文章中作者介绍了一种基于计算机驱动的方法,可以从大型天然化合物数据库中筛选出潜在的新型甜味剂。
1.介绍
作者对包含213210个天然化合物UNPD数据库(http://pkuxxj.pku.edu.cn/UNPD)进行了筛选。此工作主要分为两大模块,分别是基于数据和基于结构的方法。首先,采用数据库方法通过比较它们的化学指纹来筛选与已知甜味剂结构相似的化合物。其次,构建分类模型从结构与甜味剂相似的化合物中识别甜味化合物。此外,构建预测模型以预测所筛选的甜味化合物的甜度对数值。然后使用FAF-drugs4和pkCSM分析模型筛选分子的毒性和口服生物利用度。最后,用一种分子对接方法来首先识别新型潜在的甜味剂。整个文章的框架如图1所示。
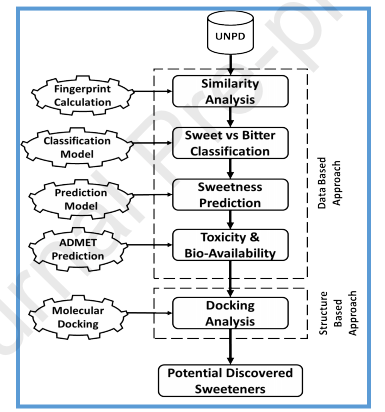

图1:用于虚拟筛选以识别新甜味剂的框架。框架由几种模型组成,这些模型分为基于数据的模块和基于结构的模块。
2. 结果
2.1相似性分析
作者比较了UNPD和478种甜味剂的化学空间(去除Tanimoto系数小于0.65的天然分子)。并且基于1024位的标准指纹,发现包含213210种天然化合物的数据库UNPD,只有10184种化合物与478种已知甜味剂在结构上相似。
2.2分类模型
首先,作者使用八个描述符(BCUT,Estate,pi-path count,chipath,自相关,拓扑化学原子,MOE电荷,VSA描述符)和随机森林算法构建模型。训练和测试集的MCC值分别为0.815和0.785,表明构建的二分类模型适用于甜味和苦味的区分。对于分类模型,获得了准确性(训练集为0.897,测试集为0.865),敏感性(训练集为0.912,测试集为0.891)和特异性(训练集为0.904,测试集为0.818)。
其次,作者通过分类模型对10184个分子进行进一步筛选,基于预测置信度大于0.7,则该分子为甜类的原则,筛选出1924个甜味化合物。
2.3预测模型
在这里,作者构建了一种机器学习模型来计算分子的相对甜度。模型获得的对数(RS)值与实验对数(RS)值具有很好的相关性。训练集和测试集的R2分别为0.845和0.798。结果如图2所示。
作者发现,疏水相互作用,静电相互作用,分子的形状和大小在甜味感知中起着至关重要的作用。

图2:训练集和测试集的实验值与模型预测对数(RS)值的图。
2.4分子对接
作者通过分子对接发现,不同类别甜味剂的结合能与其甜味呈负相关,如图3所示。评估的478种甜味剂与T1R2-T1R3的相关系数r为-0.58。观察到,两个圆圈区域是异常值,一个区域的甜度指数低,通常由糖和多元醇组成,另一区域的天然高甜度甜味剂具有出乎意料的高结合能。该组主要由萜烯(紫苏碱,甜菊糖苷等)和多酚(类黄酮,异香酚类衍生物等)组成。

图3:不同类型甜味剂的结合能(kcal / mol)与甜度指数log(RS)的关系图。给出了结合能(BE)和对数(RS)之间的关系。分子的离群区域被包围。
2.5新型甜味剂的识别
作者通过上述操作得到1354个具有高甜度的化合物。进一步的毒理学分析得到60个无毒且有不错的口服利用度的化合物。对筛选的60个分子的结合构象和结合能通过使用上述方案的分子对接计算。研究表明,这些分子与萜烯和多酚的结合能也很高,如图4所示。大多数鉴定出的天然高效甜味剂属于萜烯类,少于7%的分子是多酚或苯丙烷。

图6:不同种类甜味剂的结合能(kcal / mol)与甜度指数对数(RS)的关系图。新的分子被包围。
参考文献
- l Goel A, Gajula K, Gupta R, et al. In-silico screening of database for finding potential sweet molecules: A combined data and structure based modeling approach – ScienceDirect[J]. Food Chemistry, 2020.
关注我们

原创文章,作者:ifyoung,如若转载,请注明出处:https://www.drugfoodai.com/model-sweet.html
注意:本站原创指的是原创编译,并不主张对所介绍的工作的版权,如有侵权,请联系删除!
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 相关推荐
-
豆类中毛豆的理化特性及基于光谱学的机器学习方法在最佳采收期预测中的应用
今天介绍一篇由弗吉尼亚理工大学食品科学与技术系Dajun Yu等人于2021年8月发表于Food Chemistry的一篇文章。文中通过研究毛豆发育过程中理化性质的变化,并应用基于光谱学的机器学习(ML)技术来确定毛豆最佳采收期。对R5(始籽)、R6(满粒)和R7(初熟)生长期采收的毛豆进行了理化特性分析,并用手持式分光光度计测量了豆荚的光谱反射率(360-740nm)。根据毛豆样品的特征特性将其采收期分为“早期”、“准备期”和“晚期”,使用豆荚的光谱反射率的机器学习方法随机森林进行分类,研究结果证明该方法可以确定毛豆的最佳采收期。
-
基于人工智能的黄油品种识别作为食品掺假检测的研究范例
今天给大家介绍一篇由Gokce Iymen等人近期发表在Innovative Food Science and Emerging Technologies上的一篇文章。文章介绍了基于人工智能工具,利用简单的声音振动来识别食品中的掺假。
-
基于比色条形码组合和深度卷积神经网络的便携式食品新鲜度预测平台
今天介绍一篇来自江南大学,于2020年底发表在Advanced Materials上的一篇论文。该研究将可交叉反应的比色条形码组合和深度卷积神经网络(DCNN)结合在一起,形成了一个用于监控肉类新鲜度的系统,总体准确性为98.5%。
-
中国科学院上海技术物理研究所万雄课题组:基于β-胡萝卜素拉曼光谱定量检测的橄榄油鉴别
采用激光共聚焦拉曼技术与基于DFT的拉曼光谱相结合,准确分析了植物油的成分,并识别出低成本的仿制橄榄油。
-
Bioresource Technology:智能化的方法来可持续管理和利用食物废弃物
撰文:王雪洁 编辑:肖冉 介绍 今天介绍一篇由Zafar Said等人于2023年3月在线发表在Bioresource Technology(IF=11.89)上的文章。这篇文章主…
-
机器学习模型预测有机污染物的作物根系浓度因子
今天分享一篇近期由耶鲁大学医学院遗传学系的FengGao等人发表在Journal of Hazardous Materials (IF: 14.224)上的文章。
-
一种基于机器视觉系统和深度学习的检测姜黄粉中掺假的新方法
今天介绍一篇最近由伊朗阿达比大学生物系统工程系Ahmad Jahanbakhshi等人发表在COMPUT BIOL MED (Q1, IF: 4.589)期刊上的文章。文章研究中,采用改进的卷积神经网络(CNN)对姜黄粉末图像进行分类,以检测参假。结果表明,计算机视觉,特别是与深度学习(DL)相结合,可以成为评价姜黄粉质量和检测参假的一种有价值的方法。
-
FRONT NUTR:通过化学信息学方法从蓝莓中计算筛选抗阿尔茨海默病的新神经保护成分
今天介绍一篇由中南林业科技大学张琳教授团队发表的一篇研究性论文,由肖冉等人于2022年12月发表在国际营养学TOP期刊Frontiers in Nutrition上(JCR:Q1 IF:6.59)。本文尝试设计一种基于化学信息学方法这种有效的智能筛选模式,从蓝莓中寻找抗阿尔茨海默病(AD)的新型有效成分,并通过实验验证了预期成分的生物活性。该方法集成了先进的人工智能和化学信息学方法,实现了对所有成分的逐步分析和过滤。最后,获得了预期的新化合物氯化锦葵色素-3-O-半乳糖苷(Ma-3-gal-Cl)。这篇文章采用的创新性方法为。这项工作采用的筛选策略能够为研究者从天然产物和食物中筛选活性成分提供新的参考。
-
展望:大数据和机器学习有助于推进营养流行病学
今天介绍一篇由Morgenstern Jason D等人于2021年在Advances in Nutrition上发表的文章。营养流行病学领域面临着测量误差、饮食复杂,和残余混杂所带来的挑战。本文的目的是强调大数据和机器学习的发展如何帮助应对这些挑战。